MARTINI - MLAI pipeline prepare module
Prepare ADaM data for Machine Learning
Maike Ahrens, Sebastian Voss
Source:vignettes/prepare-adam-data.Rmd
prepare-adam-data.Rmd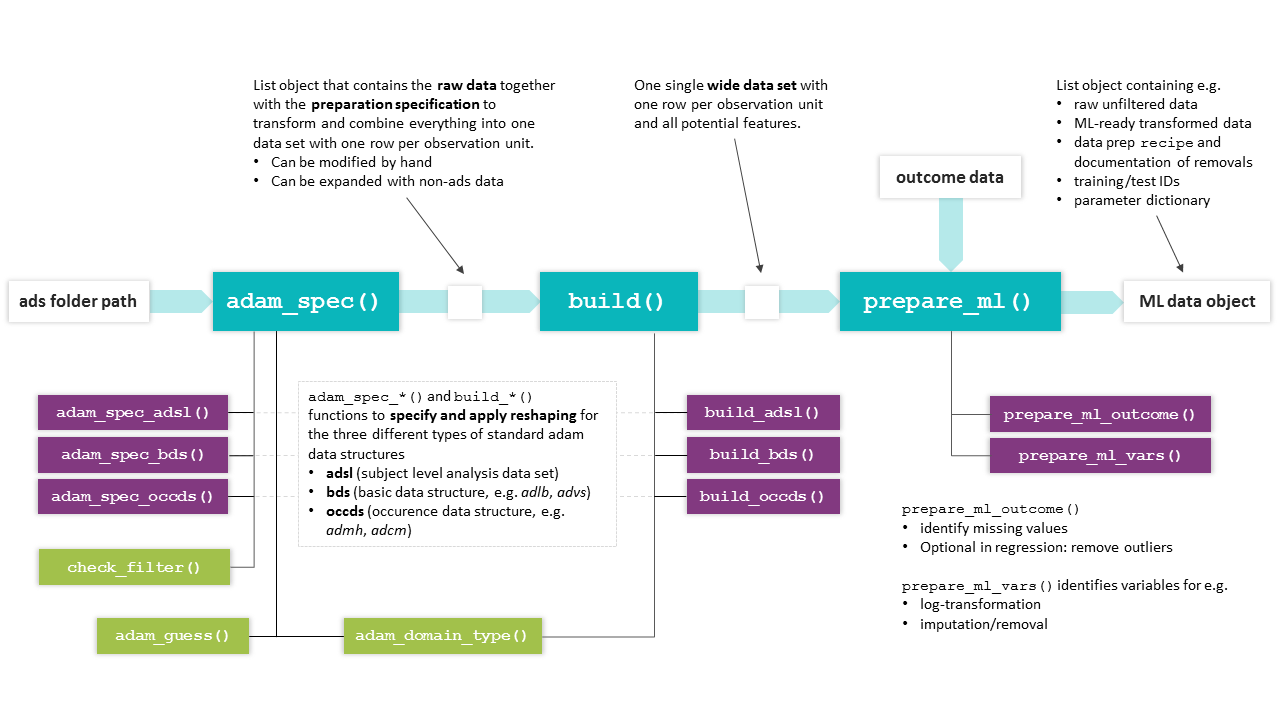
Scope
The MARTINIprep package is the first part of the BMDI
MARTINI pipeline, which aims at assessing the relation of baseline
information from clinical domains with a given outcome.
MARTINIprep provides a convenient framework to gather
information from different clinical data sets and combine them into a
machine-learning ready data set. The output is meant to be used with
packages modtune and report, both included in
the MARTINI meta package.
The preparation workflow is handled by the three main functions of
MARTINIprep
adam_spec()ads_build()prepare_ml()
The package was developed in the clinical context which is reflected in the default settings and specifics of both main and helper functions. This vignette is solely focused on the standard clinical setting. However, the functions may also be used with more general data sets, please refer to the individual help pages for full details.
Main functions
adam_spec()
In the setting of clinical studies, we assume that a set of analysis
data sets (ads) is stored in .sas7bdat format in a single
folder path.
After specifying the folder location
path <- 'path/to/sasfiles'the command
ads_spec <- adam_spec(path)will
- identify data sets that can be processed automatically (by matching file names against an internal library). In case a particular domain is not included in the current list, please get in touch with the package maintainers.
- create a preprocessing specification in the form of a list, where each entry describes the required information to extract relevant information from a particular data set and reshape the data into wide format. The structure of these entries depends on the type of data set (adsl/bds/occds).
- generate md5 checksums
- check applicability of provided filters
In general, this automatically created specification may be used with the subsequent workflow, however, in practice, it will be modified by the user to match specific requirements.
Important parameters
Filters
The filter argument takes a character vector of valid
filter expressions. A filter will be applied to a particular data only
if its application yields a non-empty tibble (i.e. no error thrown, at
least one row is selected). Common filters may be based on visit or
treatment information, as well as flags indicating analysis sets.
The pre_study argument allows to conveniently reduce
e.g. medical history records to data that is available at baseline based
on the study day.
filters <- c(
"AVISIT == 'Baseline'",
"ITTFL == 'Y'",
"!is.na(TRT01A)"
)
ads_spec <- adam_spec(
path,
filter = filters,
pre_study = TRUE
)attach data
In order to create a data set specification, the data set has to be
read first which may take a considerable amount of time for large files.
For a more time-efficient usage, the data sets may be stored directly in
the ads_spec object from where the actual execution of the
preparation will be conducted.
In the current implementation, if changes to any of the data sets shall be made (see below), all data sets have to be attached.
ads_spec <- adam_spec(
path,
attach_data = TRUE
)Manual adaptations
The most common adaptations of an automatically generated spec object are the removal of particular variables and adjustment of factor level order and/or names.
remove columns (subject level)
We’ll demonstrate removal of variables using the example of adsl.
The variables to be selected from the adsl data set are
stored in ads_spec$adsl$select. Simply remove column names
from this entry to discard particular variables. Reasons for exclusion
may include but are not limited to:
- post baseline information (MARTINI pipeline aims at assessing the relation of baseline information with a given outcome. Consequently, outcome-related information should be removed (e.g. death flag))
- variables are available in both a continuous as well as categorical version (e.g. age, BMI, weight)
- different groupings based on the same variable (e.g. country group)
ads_spec$adsl$select <- ads_spec$adsl$select %>%
# remove post baseline information
setdiff(c('DEATHFL')) %>%
# categoricals with a continuous version
str_subset("AGEGR|BMIGR|WEIGGR|RACEGR", negate = TRUE)
# several grouped versions available
str_subset("CNTYGR[3-7]") %>% If additional variables shall be included in the analysis that are not extracted from the processed sas data sets
factor level order
In order to change factor level names and/or their order, update the
factor_levels entry of the corresponding data set in the
spec object. The information is stored in a list, where the
entries are named after the column containing the factor levels
(i.e. TRT01A instead of TRT01AN). This is often used to specify the
order of the treatment variable to control the order in the result
presentation.
# change order of treatment levels
ads_spec$adsl$factor_levels$TRT01A <- ads_spec$adsl$factor_levels$TRT01A %>% na.exclude %>% rev()change data
If any manual changes need to be made to the data sets themselves,
the attach_data parameter of ads_spec() needs
to be set to TRUE. In this case, top level entry of
spec will have a data slot where the data set
is stored and used by the ads_build() function. Just like
the other slots, it may be adjusted by the user in an intermediate step
as shown in the following.
# variables that contain explicit "unknowns"
var_unknown <- ads_spec$adsl$data %>%
select_if(is.character) %>%
select_if(~{any(str_to_lower(.x) %in% c("u", "unknown"))}) %>%
colnames()
var_unknown
# recode "unknowns" as NA
ads_spec$adsl$data <- ads_spec$adsl$data %>%
mutate_at(var_unknown, ~if_else(str_to_lower(.x) %in% c("u", "unknown"), NA_character_, .x))add data
Adding data on a subject level is a special case of changing data:
overwrite the data slot of the subject level spec (again, make sure to
set attach_data = TRUE) by the extended data set.
ads_spec$adsl$data <- left_join(
ads_spec$adsl$data,
additional_data
)
Change a data set label
# change label column in admh
ads_spec$admh$label <- "MHBODSYS"ads_build()
Based on a given ads_spec object (modified or generated
fully automatically), the ads_build() function will
execute the extraction of the relevant information according to
the ads_spec entries and combine everything into a wide data set with
one row per id. This data set is the basis for the feature
matrix used later on for machine learning.
feature <- ads_build(spec)Relevant helper functions
Internally, ads_build() will call the appropriate
build_*() function for each data type (adsl/bds/occds).
While spec_bds() and spec_occds() are
rather similar in the sense that only few columns need to be extracted
and reshaped, a lot more steps are required for the preparation of
adsl.
build_ads()
- extract factor codings and rename levels to obtain valid feature names
- removal of
- datetime columns (for certain analyses)
- redundant columns (id, treatment, combined columns)
build_bds()/build_occds()
prepare_ml()
Once all potential features are available in a single data set, the
prepare_ml() function will take care of the data
preprocessing required for machine learning analysis based on the
provided outcome data (see below for recommended preparation)
ml_data <- prepare_ml(
feature,
outcome
)While each step is optional and parametrized to provide maximum flexibility to the user, default parameters were chosen carefully and may be considered appropriate for a large number of analyses.
The preprocessing includes the following steps
- splitting in training and test set (stratified by e.g. treatment)
- removal of noise
- reduction of multicollinearity by removing highly correlated variables
- log-transformation of highly skewed variables
- normalization
- imputation
- dummy coding
outcome preparation
It is recommended to prepare an outcome data set containing information on each endpoint that might be of interest including tte endpoints for which one endpoint is described by two columns (time and censor).
tte
Time to event endpoints may be analyzed as-is or as a binary version (event yes/no). Since the analysis of survival endpoints is computationally much more expensive than for classification, the latter may be used for initial runs.
Use the function binarize_tte() in order to derive
binary endpoints from a tte outcome.